R语言初学者指南-第三章
时隔半年,我终于又开始写这个博客了。 话不多说,言归正传。
第三章 访问变量和处理数据子集
在学习上一章导入数据,这一章节来学习对变量的访问和数据子集的处理。
3.1 访问数据框变量
当确认导入数据无误后,就可以按照自己的需求开始对数据进行删除部分数据,选取部分数据也就是数据子集了。 以上一章的鱿鱼数据为例。
#导入数据,并生成一个数据框
Squid <- read.table(file = "F:\\database\\RBook\\squid.txt", header = TRUE)
#查看数据框中的变量名
names(Squid)## [1] "Sample" "Year" "Month" "Location" "Sex" "GSI"3.1.1 str函数
这个函数主要是告诉我们这个Squid数据框中5个变量的属性。
str(Squid)## 'data.frame': 2644 obs. of 6 variables:
## $ Sample : int 1 2 3 4 5 6 7 8 9 10 ...
## $ Year : int 1 1 1 1 1 1 1 1 1 1 ...
## $ Month : int 1 1 1 1 1 1 1 1 1 2 ...
## $ Location: int 1 3 1 1 1 1 1 3 3 1 ...
## $ Sex : int 2 2 2 2 2 2 2 2 2 2 ...
## $ GSI : num 10.44 9.83 9.74 9.31 8.99 ...如这个结果所示,变量样本、年份、月份、位置和性别都是整数型即int,GSI是数值型即num。 为什么建议在读入数据形成数据框之后,用str函数看一下变量属性呢,因为如果在读入数据时使用了错误的分割符号:
#设定分割符号是","
Squid2 <- read.table(file = "F:\\database\\RBook\\squid.txt", dec = ",", header = TRUE)
#查看数据框中的变量属性
str(Squid2)## 'data.frame': 2644 obs. of 6 variables:
## $ Sample : int 1 2 3 4 5 6 7 8 9 10 ...
## $ Year : int 1 1 1 1 1 1 1 1 1 1 ...
## $ Month : int 1 1 1 1 1 1 1 1 1 2 ...
## $ Location: int 1 3 1 1 1 1 1 3 3 1 ...
## $ Sex : int 2 2 2 2 2 2 2 2 2 2 ...
## $ GSI : Factor w/ 2472 levels "0.0064","0.007",..: 1533 2466 2462 2445 2428 2407 2379 2308 2288 2247 ...这个时候GSI就是个因子即factor,当后续处理时,R就会报错。 后续我们将对GSI这个变量进行统计分析,值得注意的是,GSI存在于Squid这个数据框中,而没有存在于R的内存中,也就是说,在直接访问GSI的时候,R会报错。
#我注释掉了,因为不这样的话,会卡在报错那里
#GSI3.1.2 函数中的数据参数
这一节说的是啥呢,书上翻译的感觉啰嗦,其实就是在有些函数中指定数据集。毕竟你想做个线性回归什么的,你得告诉R要用到哪个数据集。
N1 <- lm(GSI~factor(Location)+factor(Year),
data = Squid)
N1##
## Call:
## lm(formula = GSI ~ factor(Location) + factor(Year), data = Squid)
##
## Coefficients:
## (Intercept) factor(Location)2 factor(Location)3 factor(Location)4
## 1.3939 -2.2178 -0.1417 0.3138
## factor(Year)2 factor(Year)3 factor(Year)4
## 1.3548 0.9564 1.2270但是有些函数的参数没有这个data=……,比如mean()函数。这个时候可以用attach()函数绑定数据集。这个可以在之后讨论。
3.1.3 $符号
上文说到有的函数没有data参数,还有一个办法是用$符号。比如:
#这个命令输出结果太多
#Squid$GSI
#于是我用了head显示前几行
head(Squid$GSI)## [1] 10.4432 9.8331 9.7356 9.3107 8.9926 8.7707还有一个方法,就是按列选择,观察数据框可以得知,GSI这个变量在Squid数据集的第6列,那么:
head(Squid[,6])## [1] 10.4432 9.8331 9.7356 9.3107 8.9926 8.7707当然,我不推荐这种方法,还得查第几列,麻烦。
3.1.4 attach函数
你看,之前提到了这个函数吧。这个函数可以把数据集添加到R的搜索路径中,这样,就可以直接访问数据集中的变量。
attach(Squid)
#直接访问变量GSI
head(GSI)## [1] 10.4432 9.8331 9.7356 9.3107 8.9926 8.7707#然后可以画图了
boxplot(GSI)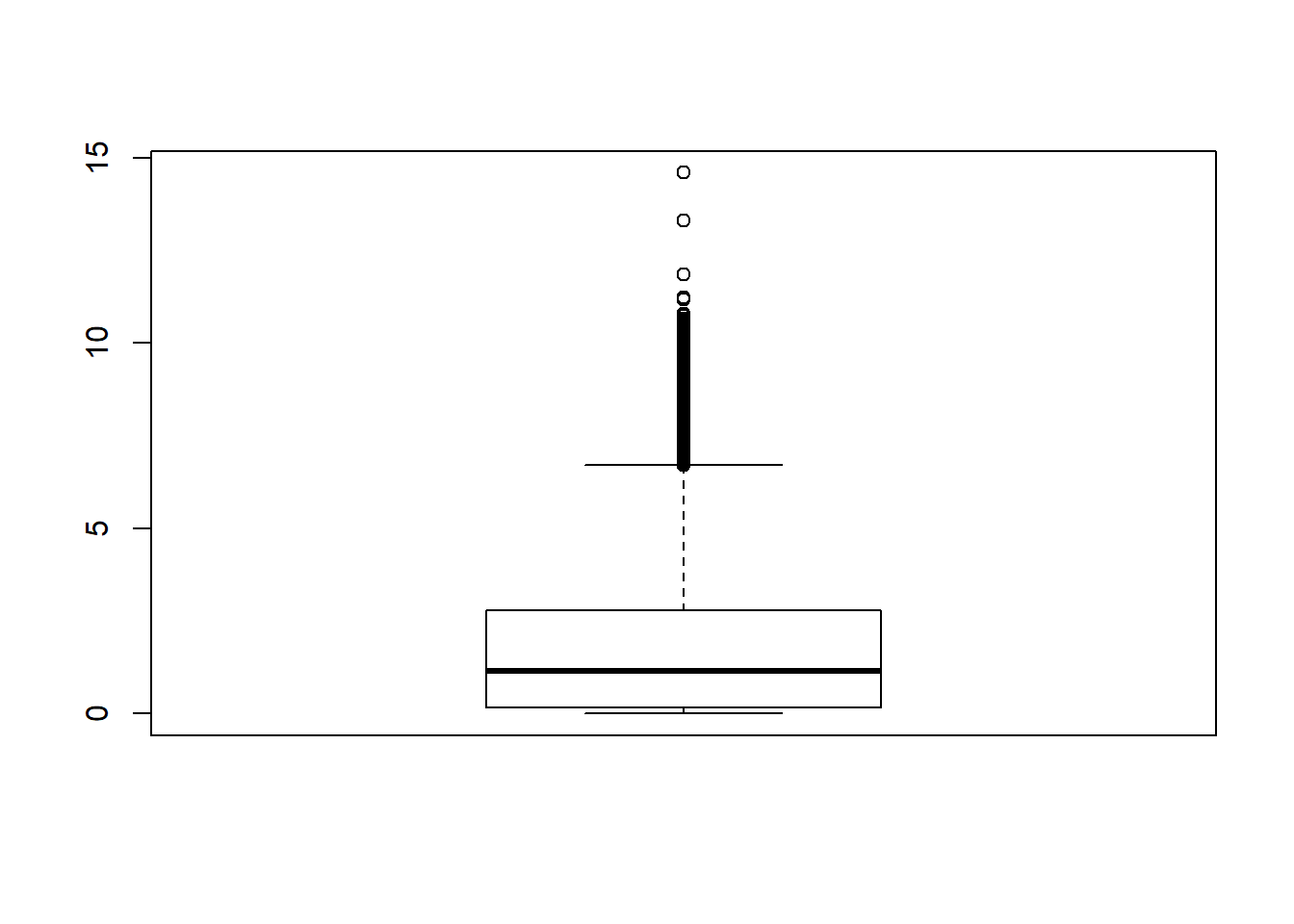
#或者运算
mean(GSI)## [1] 2.187034#当想要解绑的时候
detach(Squid)注意哈,一次绑定一个数据,当想绑定其他数据集的时候,建议最好先解绑前一个数据集,避免有同名的变量干扰。
3.2 访问数据子集
Squid这个数据集里面有个sex变量,别想多了,是性别。此时,我只想对雄性的数据进行处理,可以这么做: 首先需要知道性别是如何编码的:
unique(Squid$Sex)## [1] 2 1这里是这么表示的,雄性是1,雌性是2。 接下来访问所有的雄性数据,并存储在SquidM的数据框中:
Se1 <- Squid$Sex == 1
SquidM <- Squid[Se1,]
head(SquidM)## Sample Year Month Location Sex GSI
## 24 24 1 5 1 1 5.2970
## 48 48 1 5 3 1 4.2968
## 58 58 1 6 1 1 3.5008
## 60 60 1 6 1 1 3.2487
## 61 61 1 6 1 1 3.2304
## 62 62 1 5 3 1 3.2263下面讲下这段代码的逻辑:
- 第一行生成一个与变量Sex具有相同长度的逻辑向量Se1,如果Sex值为1,则该变量的值是TRUE,反之则为FALSE。这样的变量也被称为布尔向量,可以用来选择行。
- 接下来选择Squid中Se1等于TRUE的行,并存储在SquidM中。 至此,雄性数据选择完毕。 当然,这个代码也可以简化为:
SquidM <- Squid[Squid$Sex == 1,]
head(SquidM)## Sample Year Month Location Sex GSI
## 24 24 1 5 1 1 5.2970
## 48 48 1 5 3 1 4.2968
## 58 58 1 6 1 1 3.5008
## 60 60 1 6 1 1 3.2487
## 61 61 1 6 1 1 3.2304
## 62 62 1 5 3 1 3.2263#雌性的数据
SquidF <- Squid[Squid$Sex == 2,]
head(SquidF)## Sample Year Month Location Sex GSI
## 1 1 1 1 1 2 10.4432
## 2 2 1 1 3 2 9.8331
## 3 3 1 1 1 2 9.7356
## 4 4 1 1 1 2 9.3107
## 5 5 1 1 1 2 8.9926
## 6 6 1 1 1 2 8.7707下面讲一下布尔运算符的用法:或“|”,与“&”,非“!”。 先看一下Location变量有什么编码值:
unique(Squid$Location)## [1] 1 3 4 2然后,我只想要Location为1,2,3的数据,注意这个是并集。
Squid123 <- Squid[Squid$Location == 1 |
Squid$Location == 2 |
Squid$Location == 3,]
Squid123 <- Squid[Squid$Location != 4, ]
Squid123 <- Squid[Squid$Location < 4, ]
Squid123 <- Squid[Squid$Location <= 3, ]
Squid123 <- Squid[Squid$Location >= 1 &
Squid$Location <= 3,]以上语句都是一个意思。 额,写完才发现,我选用的是Cascadia code字体,这样的话……绝对等于也就是两个等于号是==,不等于应该是!=,而这里显示的是!=,同样的,小于等于和大于等于的形式都发生变化了。 然后,接下来,我想进一步从雄性数据集中提取出Location为1的数据集:
SquidM.1 <- Squid[Squid$Sex == 1 &
Squid$Location == 1,]3.2.1 数据排序
有的时候想对数据集根据某一个变量进行排序,从大到小啊或者从小到大,在excel中很好操作,在R中要这样:
#按月份排序
head(Squid[order(Squid$Month),], n=30L)## Sample Year Month Location Sex GSI
## 1 1 1 1 1 2 10.4432
## 2 2 1 1 3 2 9.8331
## 3 3 1 1 1 2 9.7356
## 4 4 1 1 1 2 9.3107
## 5 5 1 1 1 2 8.9926
## 6 6 1 1 1 2 8.7707
## 7 7 1 1 1 2 8.2576
## 8 8 1 1 3 2 7.4045
## 9 9 1 1 3 2 7.2156
## 11 11 1 1 1 2 6.3882
## 14 14 1 1 1 2 6.0726
## 18 18 1 1 1 2 5.7757
## 198 198 1 1 1 1 1.2610
## 204 204 1 1 1 1 1.1997
## 244 244 1 1 1 1 0.8373
## 255 255 1 1 1 2 0.6716
## 264 264 1 1 1 2 0.5758
## 271 271 1 1 3 1 0.5518
## 279 279 1 1 1 1 0.4921
## 281 281 1 1 1 1 0.4808
## 292 292 1 1 3 2 0.3828
## 302 302 1 1 1 1 0.3289
## 317 317 1 1 1 1 0.2758
## 329 329 1 1 1 1 0.2506
## 352 352 1 1 1 2 0.2092
## 373 373 1 1 1 2 0.1792
## 381 381 1 1 3 1 0.1661
## 387 387 1 1 1 2 0.1618
## 393 393 1 1 1 1 0.1543
## 394 394 1 1 1 1 0.1541因为处理的是Squid中行,所以放在了逗号的前面。也可以对一个变量进行排序:
head(Squid$GSI[order(Squid$Month)], n=30L)## [1] 10.4432 9.8331 9.7356 9.3107 8.9926 8.7707 8.2576 7.4045 7.2156
## [10] 6.3882 6.0726 5.7757 1.2610 1.1997 0.8373 0.6716 0.5758 0.5518
## [19] 0.4921 0.4808 0.3828 0.3289 0.2758 0.2506 0.2092 0.1792 0.1661
## [28] 0.1618 0.1543 0.15413.3 使用相同的标识符组合两个数据集
书里这段说的很啰嗦,其实就是在实际使用中,我们可能会导入很多个excel文件,而这些文件里面的样本是一样的。简单来说,举个例子,我用了10只小鼠,样本命名为1,2,3,……,10,第一个excel文件里记载了每只老鼠血清的甘三酯(TG)含量,第二个excel文件里,记载了每只老鼠肝脏的TG含量,这两个文件分别导入到R中,生成两个数据集,而这两个数据集,样本名是一致的。 以书中的例子而言,是这样的,Squid1.txt文件里记载了样本和对应的GSI,Squid2.txt文件里记载了样本和其他的对应变量,比如年份、月份、位置、性别。 下面来读入这两个数据:
Sq1 <- read.table(file = "F:\\database\\RBook\\Squid1.txt", header = TRUE)
Sq2 <- read.table(file = "F:\\database\\RBook\\Squid2.txt", header = TRUE)然后根据这两个数据集具有一致的样本进行合并,采用merge函数:
SquidMerged <- merge(Sq1, Sq2, by = "Sample")
head(SquidMerged, n=30L)## Sample GSI YEAR MONTH Location Sex
## 1 1 10.4432 1 1 1 2
## 2 2 9.8331 1 1 3 2
## 3 3 9.7356 1 1 1 2
## 4 5 8.9926 1 1 1 2
## 5 6 8.7707 1 1 1 2
## 6 7 8.2576 1 1 1 2
## 7 8 7.4045 1 1 3 2
## 8 9 7.2156 1 1 3 2
## 9 10 6.8372 1 2 1 2
## 10 11 6.3882 1 1 1 2
## 11 12 6.3672 1 6 1 2
## 12 13 6.2998 1 2 1 2
## 13 14 6.0726 1 1 1 2
## 14 15 5.8395 1 6 1 2
## 15 16 5.8070 1 6 1 2
## 16 17 5.7774 1 6 3 2
## 17 18 5.7757 1 1 1 2
## 18 19 5.6484 1 5 3 2
## 19 20 5.6141 1 5 1 2
## 20 21 5.6017 1 5 3 2
## 21 22 5.5510 1 6 1 2
## 22 23 5.3110 1 5 1 2
## 23 24 5.2970 1 5 1 1
## 24 25 5.2253 1 6 1 2
## 25 26 5.1667 1 6 1 2
## 26 27 5.1405 1 6 1 2
## 27 28 5.1292 1 6 1 2
## 28 29 5.0782 1 6 1 2
## 29 30 5.0612 1 6 1 2
## 30 31 5.0097 1 5 1 2这个by = “Sample”就是说,Sq1和Sq2以Sample作为相同的标识符来合并。 另外merge这个函数还有一个参数是all = TRUE/FALSE,默认情况下,这个值是FALSE,什么意思呢,就是说Sq1和Sq2的行如果有缺失值的话,就会被忽略掉,这个样本就不存在于SquidMerged数据集中,反之,则用NA填充。下面举个all = TRUE的例子:
SquidMerged2 <- merge(Sq1, Sq2, by = "Sample",
all = TRUE)
head(SquidMerged2, n=30L)## Sample GSI YEAR MONTH Location Sex
## 1 1 10.4432 1 1 1 2
## 2 2 9.8331 1 1 3 2
## 3 3 9.7356 1 1 1 2
## 4 4 9.3107 NA NA NA NA
## 5 5 8.9926 1 1 1 2
## 6 6 8.7707 1 1 1 2
## 7 7 8.2576 1 1 1 2
## 8 8 7.4045 1 1 3 2
## 9 9 7.2156 1 1 3 2
## 10 10 6.8372 1 2 1 2
## 11 11 6.3882 1 1 1 2
## 12 12 6.3672 1 6 1 2
## 13 13 6.2998 1 2 1 2
## 14 14 6.0726 1 1 1 2
## 15 15 5.8395 1 6 1 2
## 16 16 5.8070 1 6 1 2
## 17 17 5.7774 1 6 3 2
## 18 18 5.7757 1 1 1 2
## 19 19 5.6484 1 5 3 2
## 20 20 5.6141 1 5 1 2
## 21 21 5.6017 1 5 3 2
## 22 22 5.5510 1 6 1 2
## 23 23 5.3110 1 5 1 2
## 24 24 5.2970 1 5 1 1
## 25 25 5.2253 1 6 1 2
## 26 26 5.1667 1 6 1 2
## 27 27 5.1405 1 6 1 2
## 28 28 5.1292 1 6 1 2
## 29 29 5.0782 1 6 1 2
## 30 30 5.0612 1 6 1 2对比这两个组合数据集,就可以发现样本4的差别。
3.4 输出数据
上文提到我为了研究需要,将雄性鱿鱼的数据给提取出来了,并存储于SquidM的数据集中,下面,我想把这个数据集给导出来,方便我发给小伙伴们。这个时候就会用到write.table函数:
SquidM <- Squid[Squid$Sex == 1,]
write.table(SquidM, file = "F:\\database\\RBook\\MaleSquid.txt",
sep = " ", quote = FALSE,
append = FALSE, na = "NA")这样在我的文件夹中就会出现MaleSquid.txt文件。接下来,逐个讲解这个函数中各参数的含义:
- 首先要指明要导出的数据集,本例中是SquidM;
- 然后需要告诉R,我要以什么名称,在什么位置来导出这个数据集,本例中是file = “F:\database\RBook\MaleSquid.txt”;
- sep = " "是告诉R我要将数据用空格隔开,注意引号里面是空格;
- quote = FALSE是要取消字符串的引号标志,也就是标题的引号;
- append = FALSE嘛……说实话,书中说"为FALSE的话,就会打开一个新的文件,如果为TRUE,它会将变量SquidM添加到一个已经存在的文件的尾部“,我压根就没看懂,等哪天找到英文原版看一下是不是翻译的问题;
- na = “NA”的意思就简单了,SquidM的缺失值我就用NA来代替。
看一下输出来的文件长什么样:
 可以发现,按照变量名将下面的数据逐个对应后,第一列没有名字,这个就是R的问题,如果要导入到excel中,需要将第一行的所有变量名向右移一格。说到这里,我又想吐槽了,书中是这么说的:“需要把第一行转移到右侧一列”,你瞅瞅,能理解不。
可以发现,按照变量名将下面的数据逐个对应后,第一列没有名字,这个就是R的问题,如果要导入到excel中,需要将第一行的所有变量名向右移一格。说到这里,我又想吐槽了,书中是这么说的:“需要把第一行转移到右侧一列”,你瞅瞅,能理解不。
3.5 重新编码分类变量
首先说下什么是分类变量,结合例子,在前文,我们用str(Squid)命令查看了各变量的类型:
str(Squid)## 'data.frame': 2644 obs. of 6 variables:
## $ Sample : int 1 2 3 4 5 6 7 8 9 10 ...
## $ Year : int 1 1 1 1 1 1 1 1 1 1 ...
## $ Month : int 1 1 1 1 1 1 1 1 1 2 ...
## $ Location: int 1 3 1 1 1 1 1 3 3 1 ...
## $ Sex : int 2 2 2 2 2 2 2 2 2 2 ...
## $ GSI : num 10.44 9.83 9.74 9.31 8.99 ...变量Location编码为1,2,3,4,变量Sex编码为1,2。这样的变量就是分类变量,或者叫做名义变量。 虽然说这种编码方式可以将一些字符串什么的转换为数字,但是,除了数据集所有者自己,谁也不知道Location的1,2,3,4分别是什么,随着时间的变迁,所有者自己都会忘掉。 所以就有了将这类分类变量重编码的需要。
Squid$fLocation <- factor(Squid$Location)
Squid$fSex <- factor(Squid$Sex)这两句的意思是,分别在Squid数据框中生成新变量fLocation和fSex,这里用到了R中的因子概念。 那我们看一下这两个新变量长什么样子,比如说fSex:
head(Squid$fSex, n=100L)## [1] 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 1 2 2 2 2 2 2 2 2 2 2 2 2 2
## [38] 2 2 2 2 2 2 2 2 2 2 1 2 2 2 2 2 2 2 2 2 1 2 1 1 1 1 2 1 1 1 1 1 1 1 1 1 1
## [75] 1 2 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 2 1 1 1 1 1 1 1
## Levels: 1 2注意最后一行:Levels: 1 2,这告诉我们,fSex有两个水平,下面将这两个水平重新编码为“雄性”和“雌性”:
Squid$fSex <- factor(Squid$Sex, levels = c(1,2),
labels = c("M", "F"))
head(Squid$fSex, n=100L)## [1] F F F F F F F F F F F F F F F F F F F F F F F M F F F F F F F F F F F F F
## [38] F F F F F F F F F F M F F F F F F F F F M F M M M M F M M M M M M M M M M
## [75] M F M M M M M M M M M M M M M M M M F M M M M M M M
## Levels: M F这里levels = c(1,2)和labels = c(“M”, “F”)就一一对应起来,1对应M也就是雄性,2对应F也就是雌性。 现在试试用新变量fSex来做个图或者来个线性回归:
boxplot(GSI ~ fSex, data = Squid)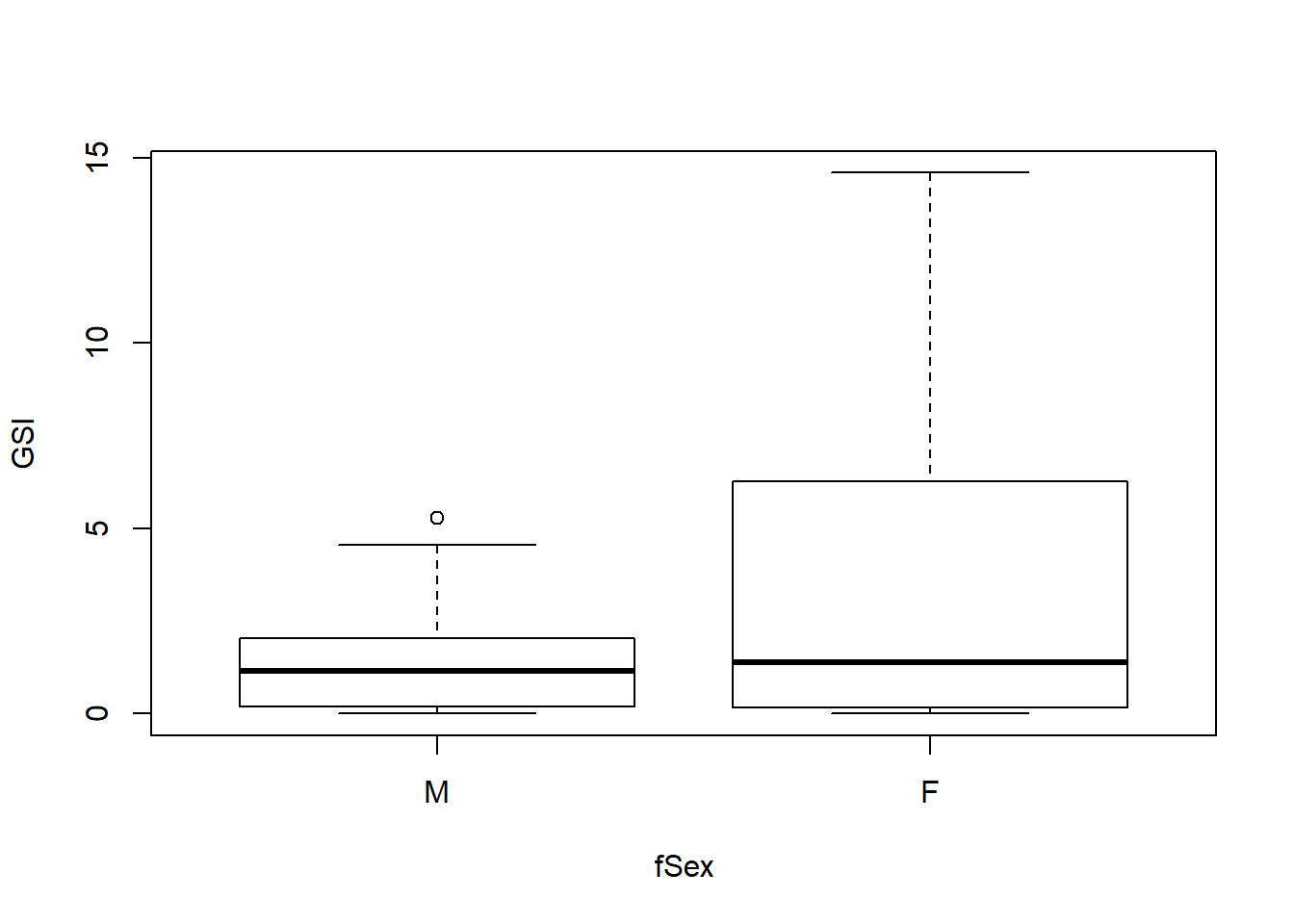
M1 <- lm(GSI ~ fSex, data = Squid)
M1##
## Call:
## lm(formula = GSI ~ fSex, data = Squid)
##
## Coefficients:
## (Intercept) fSexF
## 1.226 2.047下面,我们看一下FLocation:
head(Squid$fLocation, n=100L)## [1] 1 3 1 1 1 1 1 3 3 1 1 1 1 1 1 1 3 1 3 1 3 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
## [38] 1 1 3 1 1 1 1 3 1 1 3 1 1 1 1 1 1 1 3 1 1 1 1 1 3 1 1 1 1 1 1 1 1 1 1 1 1
## [75] 1 3 1 1 3 1 1 3 1 1 3 1 1 1 1 1 1 1 1 1 1 1 1 3 1 3
## Levels: 1 2 3 4boxplot(GSI ~ fLocation, data = Squid)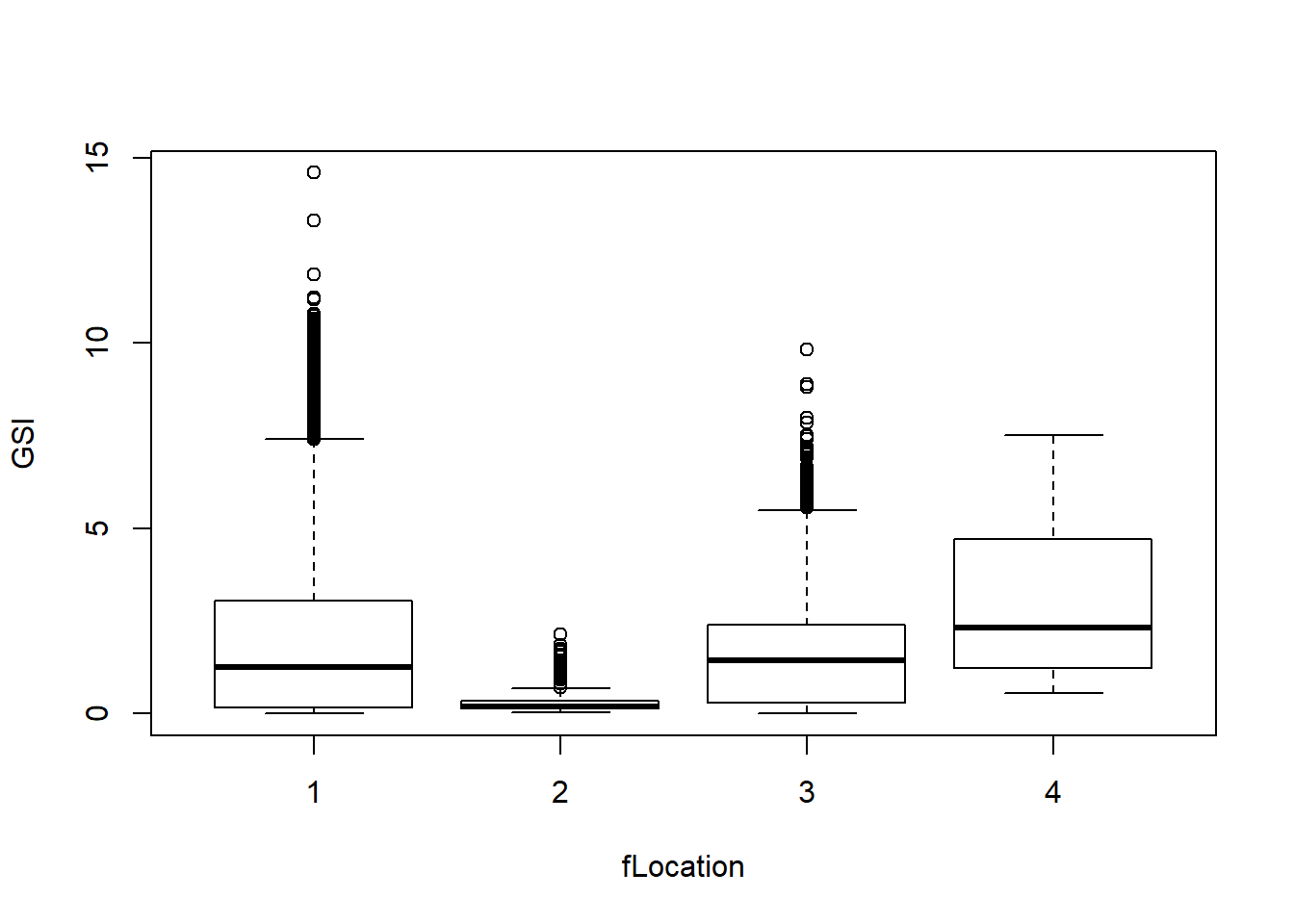
注意到,这个名义变量有四个水平,这种情况下,水平值由小到大进行排序,这意味着,在盒形图里,位置为1的数据与位置为2的数据相邻,位置为2的与位置为3的相邻等等。 但是如果我把这个默认的由小到大的排序改了呢:
Squid$fLocation <- factor(Squid$Location, levels = c(2,3,1,4))
head(Squid$fLocation, n=100L)## [1] 1 3 1 1 1 1 1 3 3 1 1 1 1 1 1 1 3 1 3 1 3 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
## [38] 1 1 3 1 1 1 1 3 1 1 3 1 1 1 1 1 1 1 3 1 1 1 1 1 3 1 1 1 1 1 1 1 1 1 1 1 1
## [75] 1 3 1 1 3 1 1 3 1 1 3 1 1 1 1 1 1 1 1 1 1 1 1 3 1 3
## Levels: 2 3 1 4此时,最后一行显示的是:Levels: 2 3 1 4,我用这个重编码过的fLocation画个图,大家看下差别:
boxplot(GSI ~ fLocation, data = Squid)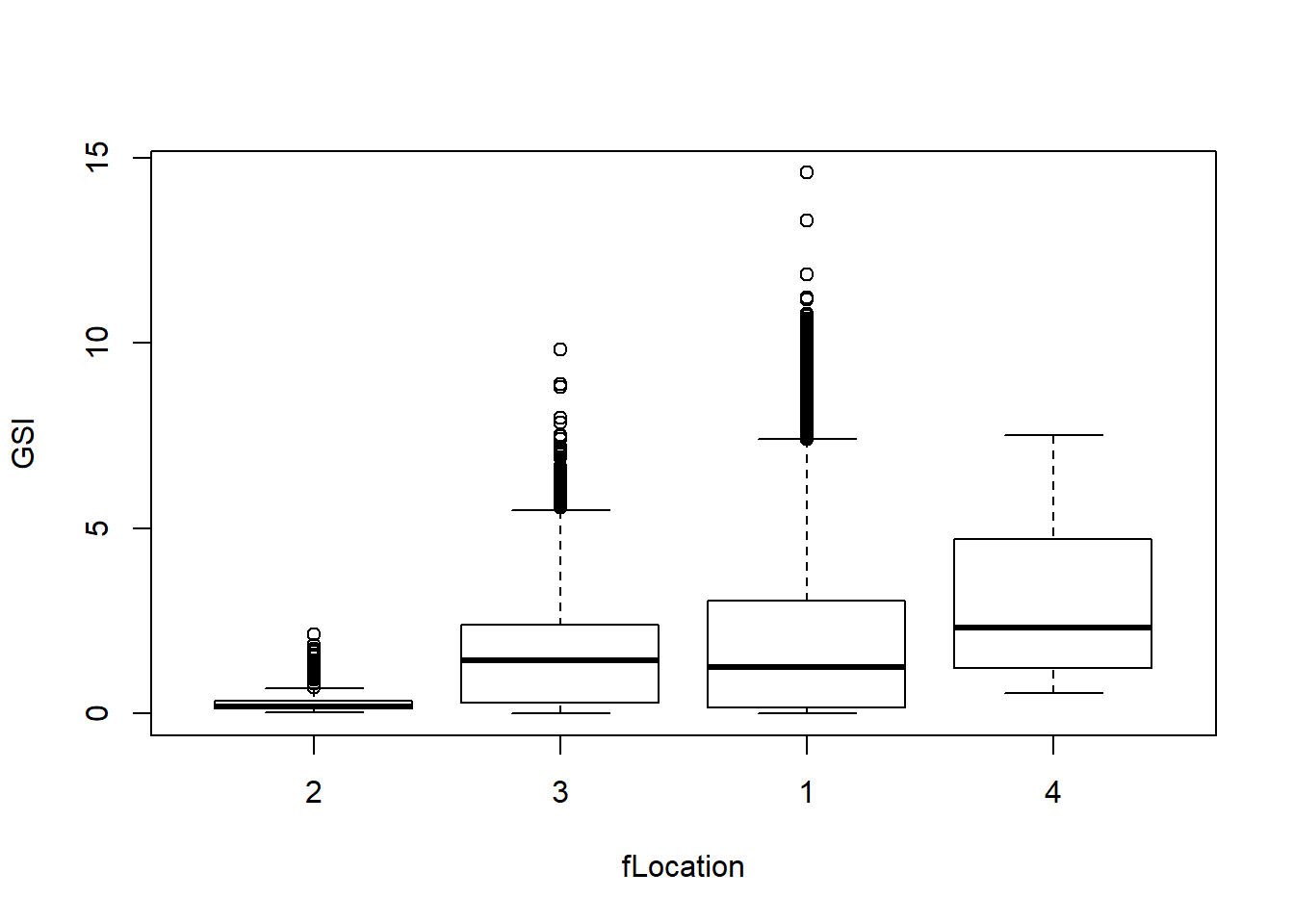
另外,在本章开始的时候选择雄性数据,我是这么做的:
SquidM <- Squid[Squid$Sex == 1,]那我想用fSex选择雄性呢,是一样的吗,答案是否定的:
SquidM <- Squid[Squid$fSex == "1",]对于fSex而言,编码数字1必须用双引号括起来,因为fSex是个因子(factor)。
3.6 本章学了哪些R函数
自己整合去。
3.7 习题
- 使用流行病学数据练习使用read.table函数并访问其中的变量: 文件为BirdFlu.xls,这是一个WHO报道的每年人类感染禽流感A(H5N1)的病例。
- 使用深海研究数据练习使用read.table函数并访问其中的变量: 这个来源于第二张的习题6,做完之后,从ISIT.xls文件载入数据。
- 使用深海研究数据练习使用write.table函数: 提取4月份并且深度超过2000米的测量数据,并输出。
- 使用深海研究数据练习使用factor函数并访问数据框中的子集: 站点1-5是2001年4月抽样,站点6-11是2001年8月抽样,站点12-15是2002年3月抽样,站点16-19是2002年10月抽样,在R里生成两个新变量确定月份和年份。